
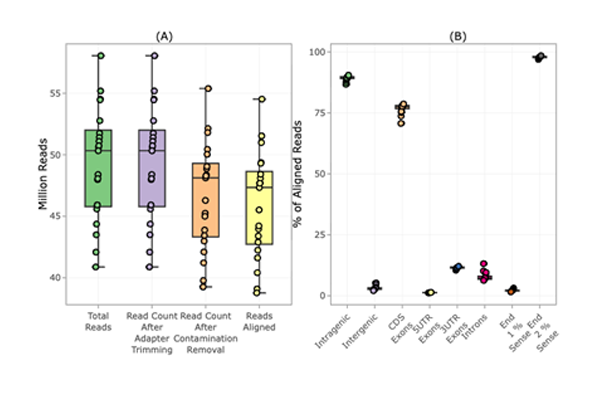
Alignment stats
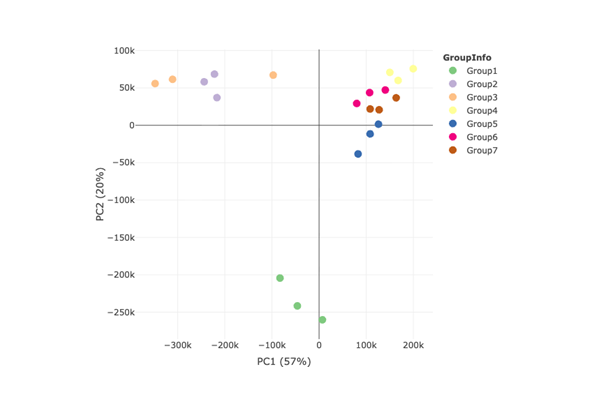
PCA Plot
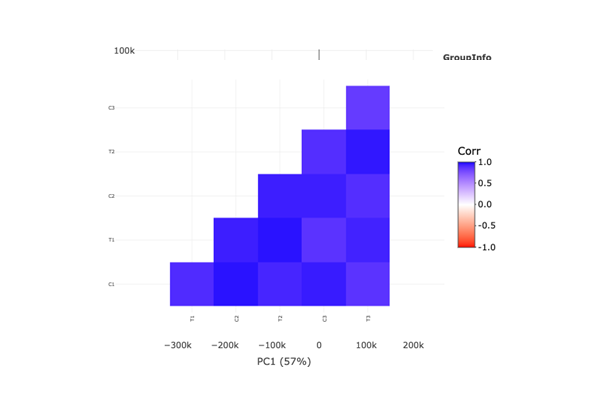
Correlation Plot
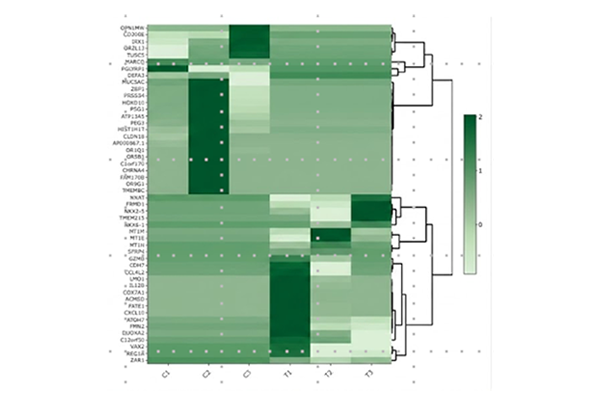
Heatmap
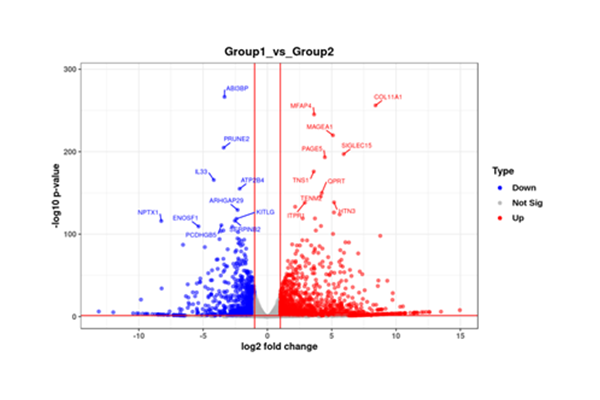
Volcano Plot
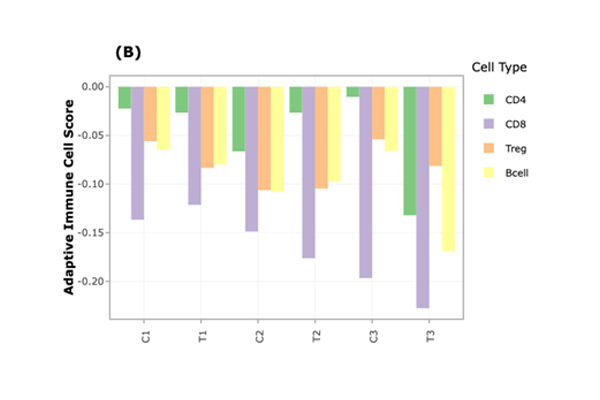
Tumor Microenvironment Analysis
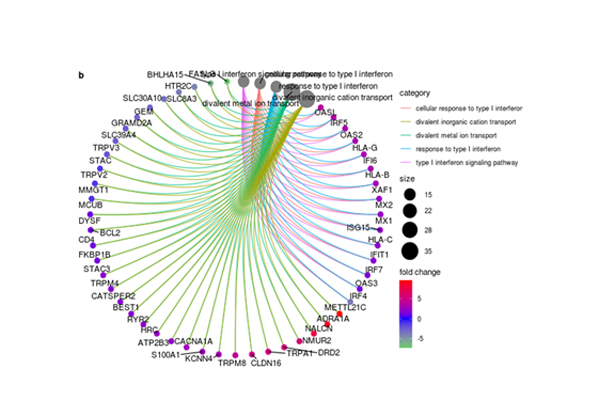
Gene Networks
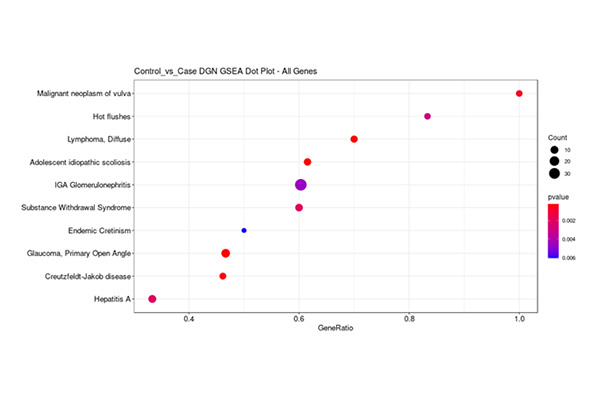
Dot Plot
![]()
![]()
![]()
![]()
![]()
![]()
![]()





Differential Gene Expression (DGE)
Quantifying transcriptional changes across experimental conditions or drug treatments.

Biomarker Discovery
Identifying unique gene signatures associated with disease states or therapeutic response.
Isoform & Splicing Analysis
Characterizing alternative splicing events and transcript-level variations.
Low-Input & Single-Cell Analysis
Extracting robust signals from precious clinical specimens and FACS-sorted populations.
Pathway & Network Analysis
Mapping expression data to biological pathways to uncover systemic insights.