

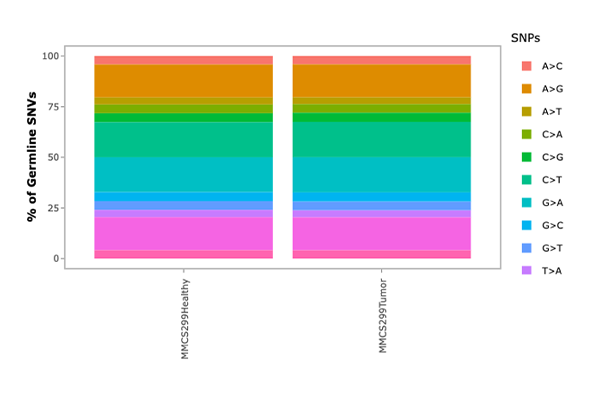
SNV & Indel Distribution
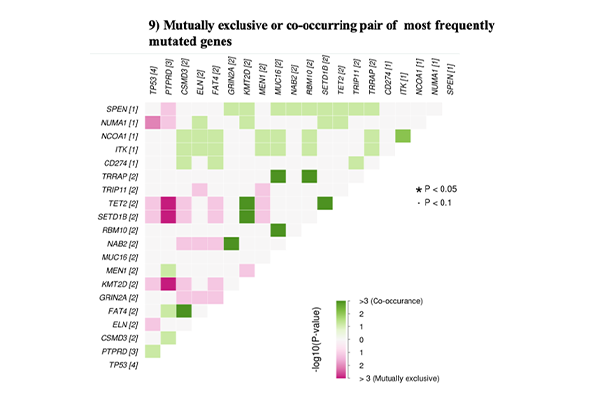
Somatic Variant Analysis
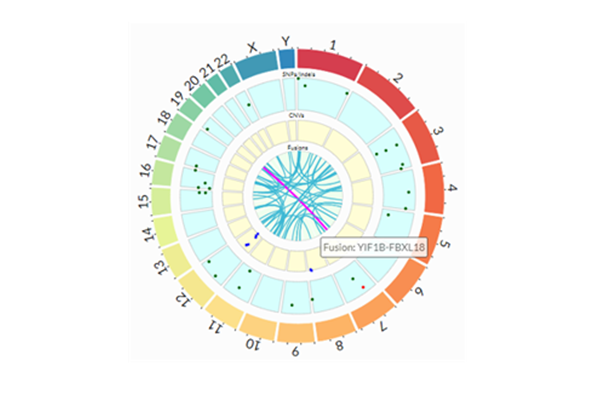
CNV & Structural Events
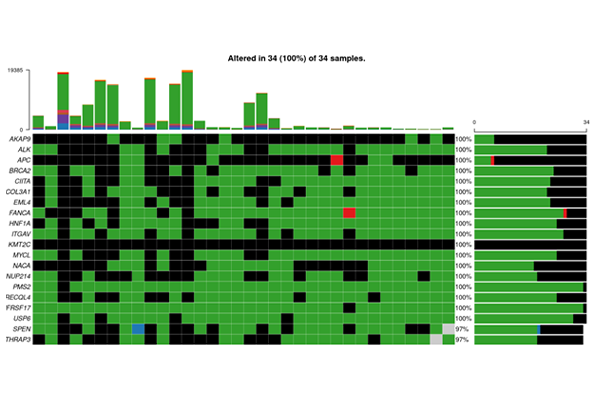
Publication-Ready Figures
![]()
![]()
![]()
![]()
![]()
![]()





Germline and Somatic Variant Detection
Identifying inherited and acquired genetic variations.
Targeted Discovery
Focusing on protein-coding regions where most disease-associated variants occur.
Rare Disease Research
Identifying causative mutations in complex phenotypes.
Oncology & Tumor Profiling
Detecting somatic variants and driver mutations.
Pharmacogenomics
Understanding how genetic variation impacts drug response.
Population Genetics
Large-scale cohort studies for genomic discovery.